All AbMole products are for research use only, cannot be used for human consumption.
In vitro: Voxtalisib (XL765) is active against class I PI3K (IC50 = 39, 113, 9 and 43 nM for p110α, β, γ and δ, respectively). XL765 also inhibits DNA-PK (IC50 = 150 nM) and mTOR (IC50 = 157 nM) but not XL-147 which shows IC50 values of > 15 μM. XL765 treatment results in decreased cell viability in 13 PDA cell lines in a dose-dependent manner. XL765, a dual-target PI3K/mTOR inhibitor, inhibits cell growth and apoptosis in many more cell lines and at lower concentrations as compared to the PI3K-selective inhibitors XL147 and PIK90. The effect can be recapitulated by using combinations of single-targeted compounds. XL765 significantly reduces phosphorylation of the mTOR targets S6, S6K, and 4EBP1, which is associated with greater apoptosis induction rather than to PI3K inhibition alone. XL765 treatment causes accumulation of autophagosomes in MIAPaCa-2 cells, and results in significant dose-dependent AVO induction and LC3-II stimulation in MIAPaCa-2 cells stably expressing a LC3-GFP construct.
In vivo: The combination of XL765 (30 mg/kg) with chloroquine (50 mg/kg) results in significant inhibition of BxPC-3 xenograft growth in mice models, while XL765 alone at the same dose has no inhibitory effect. Oral administration of XL765 results in greater than 12-fold reduction in median tumor bioluminescence compared to control and improvement in median survival in nude mice implanted intracranially with GBM 39-luc cells. XL765 in combination with temozolomide (TMZ) yields a 140-fold reduction in median bioluminescence with a trend toward improvement in median survival compared with TMZ alone.
 |
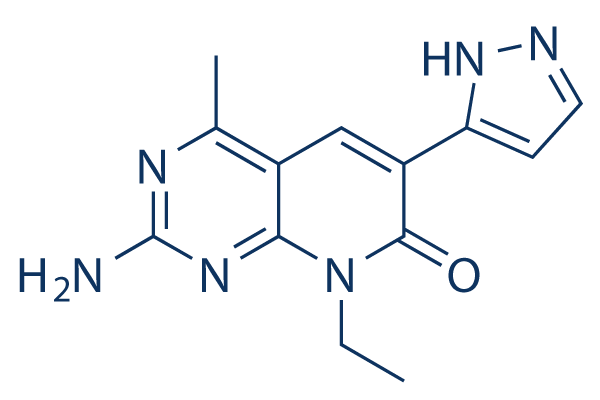
Source | Mol Cancer Ther (2014). Figure 2.XL765 |
| Method | Western immunoblotting | |
| Cell Lines | MCF7 cells | |
| Concentrations | 10 μmol/L | |
| Incubation Time | 20 h | |
| Results | XL765 was identified following optimization of a pyridopyrimidinone scaffold for in vivo PI3K/mTOR pathway inhibition and drug-like properties. |
| Cell Experiment | |
|---|---|
| Cell lines | Pancreatic cancer cell lines |
| Preparation method | Cells are treated with XL765 24 hours after plating and harvested for apoptosis or autophagy assays at 24, 48, or 72 hours after XL765 treatment. Apoptosis is determined by total percentage of annexin V-positive cells by fluorescence-activated cell sorting (FACS). Acidic vesicular organelles (AVOs) are detected in XL765-treated cells by vital staining with acridine orange. The degree of AVO formation is expressed as fold increase of acridine orange fluorescence intensity (FL3) in XL765-treated cells versus control cells. |
| Concentrations | ~10 μM |
| Incubation time | 24, 48, 72 h |
| Animal Experiment | |
|---|---|
| Animal models | Female Nu/Nu mice inoculated s.c. with BxPC-3 cells |
| Formulation | Solubilized in water/10 mM HCl |
| Dosages | 30 mg/kg |
| Administration | oral |
| Molecular Weight | 270.29 |
| Formula | C13H14N6O |
| CAS Number | 934493-76-2 |
| Solubility (25°C) | DMSO 20 mg/mL |
| Storage |
Powder -20°C 3 years ; 4°C 2 years In solvent -80°C 6 months ; -20°C 1 month |
| Related PI3K Products |
|---|
| AS605240
AS-605240 is a potent and selective inhibitor of PI 3-kinase γ (PI3Kγ) with an IC50 of 8 nM. |
| BEZ235
BEZ235 (NVP-BEZ235) is a potent dual PI3K and mTOR inhibitor of p110α, p110γ, p110δ and p110β with IC50 of 4 nM, 5 nM, 7 nM and 75 nM, respectively. |
| BKM120 (Buparlisib)
BKM120 (NVP-BKM120) is a potent and highly specific oral pan-class I PI3K inhibitor. |
| GDC-0941 (Pictilisib)
GDC-0941 is a selective and potent inhibitor of Class I PI3K, p110a IC50=0.003uM, U87MG IC50=0.95μM. |
| AS-252424
AS-252424 is a furan-2-ylmethylene thiazolidinedione as a selective ATP-competitive PI3Kγ inhibitor with IC50 with 33 nM. |
All AbMole products are for research use only, cannot be used for human consumption or veterinary use. We do not provide products or services to individuals. Please comply with the intended use and do not use AbMole products for any other purpose.
Products are for research use only. Not for human use. We do not sell to patients.
© Copyright 2010-2026 AbMole BioScience. All Rights Reserved.
