All AbMole products are for research use only, cannot be used for human consumption.
JNJ-7706621 is a novel cell cycle inhibitor that showed potent inhibition of several cyclin-dependent kinases (CDK) and Aurora kinases and selectively blocked proliferation of tumor cells of various origins but was about 10-fold less effective at inhibiting normal human cell growth in vitro. In human cancer cells, treatment with JNJ-7706621 inhibited cell growth independent of p53, retinoblastoma, or P-glycoprotein status; activated apoptosis; and reduced colony formation. At low concentrations, JNJ-7706621 slowed the growth of cells and at higher concentrations induced cytotoxicity. Inhibition of CDK1 kinase activity, altered CDK1 phosphorylation status, and interference with downstream substrates such as retinoblastoma were also shown in human tumor cells following compound treatment. Flow cytometric analysis of DNA content showed that JNJ-7706621 delayed progression through G1 and arrested the cell cycle at the G2-M phase. Additional cellular effects due to inhibition of Aurora kinases included endoreduplication and inhibition of histone H3 phosphorylation. JNJ-7706621 is a unique inhibitor regulating cell cycle progression at multiple points, suggesting that it could be useful for cell cycle analysis and therapy of various cancers, including Ewing's sarcoma.
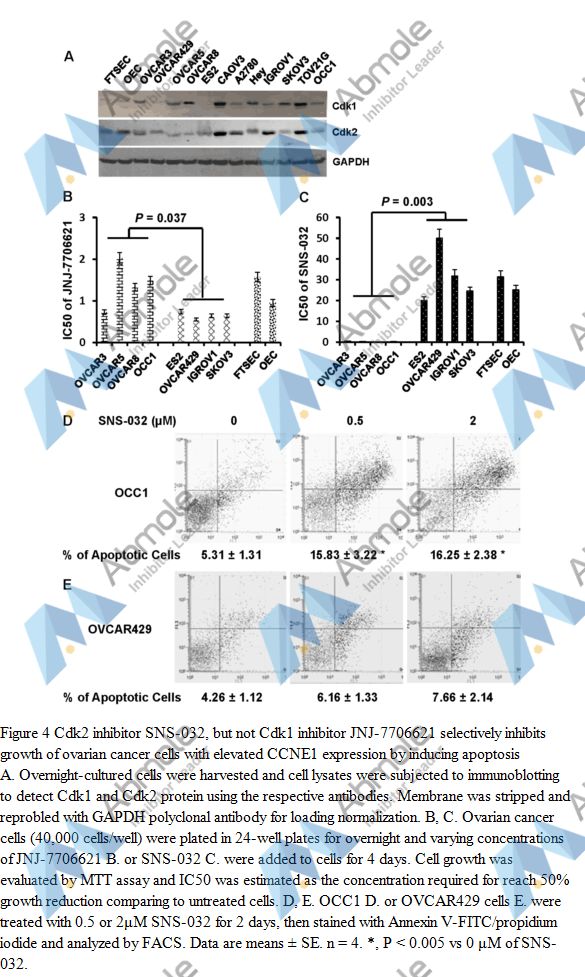 |
Source | Oncotarget (2015). Figure 4. JNJ-7706621 |
| Method | MTT assay | |
| Cell Lines | Ovarian cancer cells | |
| Concentrations | 0.5 or 2μM | |
| Incubation Time | 4 days | |
| Results | This reveals a less than 4-fold difference in the sensitivity to JNJ-7706621 between cell lines with and without CCNE1 overexpression |
| Cell Experiment | |
|---|---|
| Cell lines | HeLa cells |
| Preparation method | Colony formation assay. HeLa cells were plated at various densities in 90-mm diameter Petri dishes and exposed to JNJ-7706621 for 48 hours. Cells were then washed in PBS and incubated an additional 7 days in drug-free medium. Cells were fixed in 95% ethanol, stained with 0.5% crystal violet, and colonies containing >50 cells manually counted. |
| Concentrations | 0~3μM |
| Incubation time | 48 h |
| Animal Experiment | |
|---|---|
| Animal models | A375 melanoma human tumor xenograft model in female nu/nu mice |
| Formulation | normal saline |
| Dosages | 100 and 125 mg/kg daily |
| Administration | i.p. |
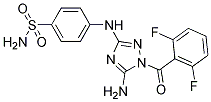
| Molecular Weight | 394.36 |
| Formula | C15H12F2N6O3S |
| CAS Number | 443797-96-4 |
| Solubility (25°C) | DMSO 69 mg/mL |
| Storage |
Powder -20°C 3 years ; 4°C 2 years In solvent -80°C 6 months ; -20°C 1 month |
| Related CDK Products |
|---|
| AT7519
AT7519 is a novel small molecule multi-cyclin-dependent (CDK) kinase inhibitor. |
| Flavopiridol
Flavopiridol (Alvocidib) is a competitive broad-spectrum CDK inhibitor with IC50 of 30,170,100 nM against CDK1, CDK2 and CDK4, respectively. |
| R547
R547 is a potent and selective ATP-competitive CDK inhibitor, with Ki values of 2 nM, 3 nM and 1 nM for CDK1/cyclin B, CDK2/cyclin E and CDK4/cyclin D1, respectively. |
| PD 0332991 HCL
PD-0332991 HCl is a highly selective inhibitor of CDK4/6 with IC50 of 11 nM/16 nM in cell-free assays, respectively. It shows no activity against CDK1/2/5, EGFR, FGFR, PDGFR, InsR, etc. |
| SCH727965 (dinaciclib)
SCH727965 (Dinaciclib) is a potent and selective CDK inhibitor with IC50 values of 1, 1, 3 and 4 nM for CDK2, CDK5, CDK1 and CDK9, respectively. |
All AbMole products are for research use only, cannot be used for human consumption or veterinary use. We do not provide products or services to individuals. Please comply with the intended use and do not use AbMole products for any other purpose.
Products are for research use only. Not for human use. We do not sell to patients.
© Copyright 2010-2026 AbMole BioScience. All Rights Reserved.
