All AbMole products are for research use only, cannot be used for human consumption.
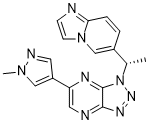
In vitro: Volitinib has exquisite kinase selectivity and excellent potency. Volitinib displays a highly selective profile across a gastric cell line panel, potently inhibiting cell growth only in those lines with dysregulated cMET (EC50 values 0.6 nM/L-12.5 nM/L). Volitinib has high membrane permeability without efflux transport across Caco-2 cell monolayer and exhibits negligible P-gp inhibition (IC50 > 17 μM). Volitinib shows no significant reversible or mechanism-based CYP inhibition in human liver microsomes, and no induction of CYP1A2 and CYP3A4 in human hepatocytes.
In vivo: In a mouse pharmacokinetic study (male ICR mice), the clearance of the compound is 4.28 L/(h·kg) and the half-time is 1.7 h. Despite its moderate oral bioavailability (F = 27.2%), the overall plasma exposure is much higher. Volitinib demonstrates dose-dependent tumor growth inhibition in a U87MG subcutaneous xenograft model. Its treatment leads to pharmacodynamic modulation of c-MET signaling and potent tumor stasis in 3/3 cMET-dysregulated GC PDX models, but has negligible activity in a GC control model. Volitinib has moderate plasma protein binding rate (60%∼70% in rat, dog, and human; 40% in mouse; 80% in monkey) and exhibits wide distribution to different organs in rat, with high exposures in liver and kidney, very low in brain, spinal cord and testis comparing to the plasma level. In PK studies in mouse, rat and dog, Volitinib shows the rapid oral absorption (Tmax<2.5 h) with high exposures and the acceptable bioavailability at 27.2%, 42.6% and 86.3%, respectively. The in vivo clearance (CL) was 11.0, 11.8 and 3.5 mL/min/kg in mouse, rat and dog, respectively, revealing a low extraction ratio. The volume of distribution in steady state (Vss) is 0.4, 1.4 and 1.4 L/kg in those species, respectively, indicating a moderate to low distribution pattern. Volitinib also displays linear pharmacokinetics (PK) in the dose ranges of 1 to 25 mg/kg in rat and 2 to 10 mg/kg in dog. Food hardly affected its PK profile in dog. In contrast, volitinib in monkey shows a notably high extraction ratio (CL=17.2 mL/min/kg) consistent with the in vitro metabolism result. Considering the rapid absorption of volitinib (Tmax=1.9 h) and moderately low distribution (Vss=0.7 L/kg), the poor oral bioavailability (1.9%) of volitinib in monkey is considered to be the result of excessive first-pass extraction. Overall, volitinib exhibits favorable preclinical PK/ADME properties.
| Cell Experiment | |
|---|---|
| Cell lines | NCI-H441 cells |
| Preparation method | NCI-H441 cells are plated at a density of 15,000 cells/well in RPMI-1640 medium with 10% FBS in 96-well plates. After incubation overnight, cells are then treated with serially diluted test compounds at 37 ℃ for 1 h. Then the medium is removed, and cells are lysed in 100 μL/well lysis buffer (1% NP-40, 20 mM Tris/pH 8.0, 137 mM NaCl, 10% glycerol, 2 mM EDTA, 1 mM activated sodium orthovanadate, 10 mg/mL Aprotinin, 10 mg/mL Leupeptin). The plates containing cell lysate are kept at -80℃ overnight. The next day, the plates are thawed on ice, mixed gently. 25 μL/well of lysates are added into the assay plates pre-coated with anti-p-Met antibody to detect p-c-Met signal. p-c-Met level is determined at 450 nm and 570 nm. |
| Concentrations | |
| Incubation time | 1 h |
| Animal Experiment | |
|---|---|
| Animal models | Female athymic mice |
| Formulation | 0.5% CMC-Na (oral); 0.25% DMSO, 10% Solutol, 10% Ethanol and 79.75% Saline (i.v.) |
| Dosages | 1.0, 2.5 and 10.0 mg/kg (oral); 2.5 mg/kg (i.v.) |
| Administration | oral administration/i.v. |
| Molecular Weight | 345.36 |
| Formula | C17H15N9 |
| CAS Number | 1313725-88-0 |
| Solubility (25°C) | DMSO 16 mg/mL |
| Storage |
Powder -20°C 3 years ; 4°C 2 years In solvent -80°C 6 months ; -20°C 1 month |
| Related c-Met Products |
|---|
| Zurletrectinib
Zurletrectinib (ICP-723) is a potent tyrosine kinase inhibitor. Zurletrectinib serves as an antineoplastic agent. Zurletrectinib can used for research of TRK-mediated related diseases. |
| ABN401
ABN401 is a highly potent and selective ATP-competitive c-MET inhibitor with an IC50 value of 10 nM. |
| (Z)-Semaxanib
(Z)-Semaxanib is a potent tyrosine kinase inhibitor. |
| MET kinase-IN-2
MET kinase-IN-2 is a potent, selective, orally bioavailable MET kinase inhibitor with an IC50 of 7.4 nM. |
| BMS-817378
BMS-817378 is a potent and selective inhibitor of MET with IC50 of 1.7 nM. |
All AbMole products are for research use only, cannot be used for human consumption or veterinary use. We do not provide products or services to individuals. Please comply with the intended use and do not use AbMole products for any other purpose.


Products are for research use only. Not for human use. We do not sell to patients.
© Copyright 2010-2024 AbMole BioScience. All Rights Reserved.
