All AbMole products are for research use only, cannot be used for human consumption.
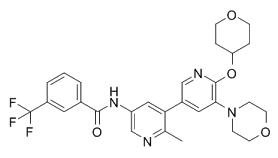
RAF709 is a potent inhibitor of B/C RAF kinase with almost equivalent IC50 values of 0.4 nM for B-RAF and C-RAF.
In cellular assays, the dose−response of pMEK and pERK are measured in Calu-6 cells with EC50 of 0.02 and 0.1 μM with minimal paradoxical activation and inhibition of proliferation with EC50 of 0.95 μM. RAF709 stabilizes BRAF−CRAF dimers with an EC50 of 0.8 μM. Of the 456 kinases tested, RAF709 shows a high level of selectivity, demonstrating greater than 99% on-target binding to BRAF, BRAFV600E, and CRAF at 1 μM and very few off-targets with DDR1 (>99%), DDR2 (86%), FRK (92%), and PDGFRb (96%), the only kinases with binding >80% at 1 μM.
In vivo: In pharmacokinetic experiments, RAF709 has moderate clearance in mouse (35 mL/min/kg) and dog (14 mL/min/kg) and high clearance in rat (50 mL/min/kg). Cmax in mouse (1 μM), dog (0.5 μM), and rat (0.5 μM) reach pharmacologically active concentrations, and acceptable oral availability is observed in mouse (68%), rat (24%), and dog (48%). In the Calu-6 xenograft nude mouse model, treatment with RAF709 results in dose-dependent antitumor activity with 10 mg/kg being subefficacious (%T/C = 92%), 30 mg/kg resulted in measurable antitumor activity (% T/C = 46%), and 200 mg/kg resulted in mean tumor regression of 92%, while the same high dose is not efficacious in the PC3, KRAS WT mode.
| Cell Experiment | |
|---|---|
| Cell lines | HCT116 cells |
| Preparation method | HCT116 cells are treated with DMSO or RAF709 at indicated concentrations for 1 hour; 1 mmol/L dabrafenib treatment is included for comparison. BRAF/CRAF dimerization is assessed by immunoprecipitating BRAF or CRAF, followed by Western blot analysis of BRAF and CRAF. Levels of pMEK and pERK in whole-cell lysates (WCL) are determined by Western blot analysis. GAPDH level is included as a loading control. |
| Concentrations | 0.1, 1, 10 μM |
| Incubation time | 1 hour |
| Animal Experiment | |
|---|---|
| Animal models | Calu-6 model (tumor bearing mice) |
| Formulation | 20% Captisol |
| Dosages | 10, 30, or 200 mg/kg |
| Administration | oral administration |
| Molecular Weight | 542.55 |
| Formula | C28H29F3N4O4 |
| CAS Number | 1628838-42-5 |
| Solubility (25°C) | DMSO 100 mg/mL Ethanol 100 mg/mL |
| Storage |
Powder -20°C 3 years ; 4°C 2 years In solvent -80°C 6 months ; -20°C 1 month |
| Related Raf Products |
|---|
| Sorafenib-d3
Sorafenib-d3 (Donafenib) is a deuterium-labeled Sorafenib, it is the first deuterium-generation tumor suppressor small molecule. Sorafenib is a multikinase inhibitor IC50s of 6 nM, 20 nM, and 22 nM for Raf-1, B-Raf, and VEGFR-3, respectively. |
| XL-281
XL-281 (BMS-908662) is an orally active inhibitor for RAF kinase, with IC50s of 2.6, 4.5 and 6 nM, for CRAF, B-RAF, and B-RAFV600E, respectively. |
| B-Raf IN 2
B-Raf IN 2 is a potent and selective BRAF inhibitor. |
| Brimarafenibum
Brimarafenibum (Brimarafenib) is a rapidly accelerated fibrosarcoma (Raf) kinase inhibitor, wtih antineoplastic effect. |
| Belvarafenib TFA
Belvarafenib TFA (HM95573 TFA) is a potent and pan RAF (Rapidly Accelerated Fibrosarcoma) inhibitor, with IC50s of 56 nM, 7 nM and 5 nM for B-RAF, B-RAFv600E and C-RAF respectively. |
All AbMole products are for research use only, cannot be used for human consumption or veterinary use. We do not provide products or services to individuals. Please comply with the intended use and do not use AbMole products for any other purpose.


Products are for research use only. Not for human use. We do not sell to patients.
© Copyright 2010-2024 AbMole BioScience. All Rights Reserved.
