All AbMole products are for research use only, cannot be used for human consumption.
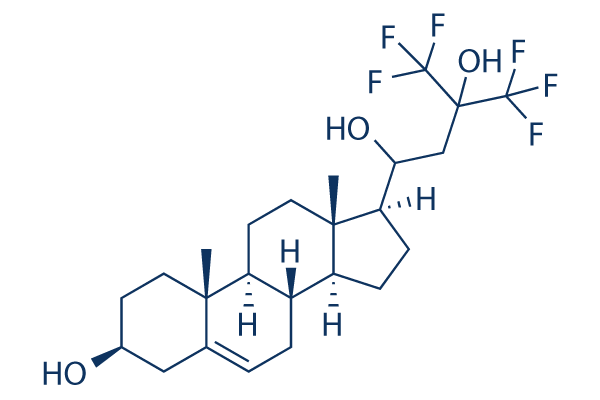
In vitro: NSC12 inhibits FGF-dependent tumor growth, angiogenesis, and metastases. NSC12 does not affect FGF2/heparin interaction, whereas it inhibits the binding of FGF2 to the immobilized receptor (ID50 ∼30 μM). NSC12 interferes with FGF2/FGFR1 interaction without affecting the ability of the growth factor to interact with heparin or HSPGs. NSC12 also binds immobilized FGF3, FGF4, FGF6, FGF8, FGF16, FGF18, FGF20, and FGF22 with Kd values ranging between ∼16 and ∼120 μM. NSC12 may act as a multi-FGF trap by interacting with all members of the canonical FGF subfamilies. NSC12 hampers FGF23-mediated FGFR1 activation in Klotho-expressing Chinese hamster ovary (CHO) cells. Treatment with NSC12 causes the reduction of the S phase of the cell cycle in all tumor cell lines but LLC cells, in which an accumulation in the S phase is observed. NSC12 inhibits FGFR1, FGFR2, FGFR3, and FGFR4 phosphorylation in CHO cell transfectants. NSC12 inhibits the proliferation of various FGF-dependent murine and human cancer cell lines with no inhibitory effect on HCC827 cancer cells that harbor a tumor-driving mutation of the EGFR TK domain and on FGF-independent cancer cell lines. In vivo: Parenteral and oral delivery of NSC12 inhibits FGFR activation, tumor growth, angiogenesis, and metastasis in FGF-dependent murine and human tumor models. NSC12 causes a significant decrease of tumor weight, tumor cell FGFR1 phosphorylation and proliferation, and tumor CD31+ neovascularization at all the doses tested in the animal models.
| Cell Experiment | |
|---|---|
| Cell lines | KATO Ⅲ cells |
| Preparation method | KATO Ⅲ cells are plated at 104 cells/well in 96 well-plates in RPMI medium plus 1% FBS. After 24 hr cells are treated with different FGFs (30 ng/ml) in the absence or presence of an optimal dose of NSC12 (1.0 or 3.0 μM) or NSC21. After 72 hr the MTT assay is performed according to manufacturer’s instructions. The optical density (OD) is determined using a plate reader at a test wavelength of 595 nm and a reference wavelength of 630 nm. |
| Concentrations | 1.0 or 3.0 μM |
| Incubation time | 72 h |
| Animal Experiment | |
|---|---|
| Animal models | C57BL/6 mice |
| Formulation | DMSO |
| Dosages | from 2.5 to 10 mg/kg |
| Administration | i.p. |
| Molecular Weight | 484.52 |
| Formula | C24H34F6O3 |
| CAS Number | 102586-30-1 |
| Solubility (25°C) | 96 mg/mL in DMSO |
| Storage |
Powder -20°C 3 years ; 4°C 2 years In solvent -80°C 6 months ; -20°C 1 month |
| Related FGFR Products |
|---|
| SNIPER(TACC3)-11
SNIPER(TACC3)-11 is a potent FGFR3-TACC3 degrader. |
| LY-3076226
LY-3076226 is an antibody drug coupling (ADC) with a cleavable linker targeting FGFR2. |
| Aprutumab ixadotin
Aprutumab ixadotin is an antibody drug coupling (ADC) with a non-cleavable linker that targets FGFR2. |
| Gunagratinib
Gunagratinib is an orally active, potent, selective, and irreversible fibroblast growth factor receptor (pan-FGFR) inhibitor for use in cancer research. |
| SAR-442501
SAR-442501 is a novel anti-FGFR3 monoclonal antibody for studies related to achondroplasia (ACH). |
All AbMole products are for research use only, cannot be used for human consumption or veterinary use. We do not provide products or services to individuals. Please comply with the intended use and do not use AbMole products for any other purpose.


Products are for research use only. Not for human use. We do not sell to patients.
© Copyright 2010-2024 AbMole BioScience. All Rights Reserved.
