All AbMole products are for research use only, cannot be used for human consumption.
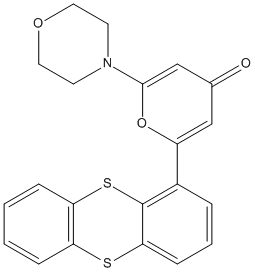
KU-55933 is a cell-permeable, potent, selective and ATP-competitive inhibitor of ATM (Ataxia telangiectasia mutated), a serine/threonine protein kinase, that exhibits an IC50 of 13 nmol/L and a Ki of 2.2 nmol/L. KU-55933 shows specificity with respect to inhibition of other phosphatidylinositol 3'-kinase-like kinases. Cellular inhibition of ATM by KU-55933 was demonstrated by the ablation of ionizing radiation-dependent phosphorylation of a range of ATM targets, including p53, gammaH2AX, NBS1, and SMC1. KU-55933 did not show inhibition of UV light DNA damage induced cellular phosphorylation events. Exposure of cells to KU-55933 resulted in a significant sensitization to the cytotoxic effects of ionizing radiation and to the DNA double-strand break-inducing chemotherapeutic agents, etoposide, doxorubicin, and camptothecin. Inhibition of ATM by KU-55933 also caused a loss of ionizing radiation-induced cell cycle arrest. By contrast, KU-55933 did not potentiate the cytotoxic effects of ionizing radiation on ataxia-telangiectasia cells, nor did it affect their cell cycle profile after DNA damage. We conclude that KU-55933 is a novel, specific, and potent inhibitor of the ATM kinase.

Cellular Signalling. 2024 Jul 26.
Early inhibition of the ATM/p53 pathway reduces the susceptibility to atrial fibrillation and atrial remodeling following acute myocardial infarction
KU-55933 purchased from AbMole
 |
Source | J Neurooncol (2012). Figure 2. KU-55933 |
| Method | clonogenic assay | |
| Cell Lines | U251 and U87 malignant glioma cell lines | |
| Concentrations | 10 microM | |
| Incubation Time | 24 hours or 72 hours | |
| Results | U87 cells also were sensitized by KU-55933 treatment, although the extent of sensitization was less profound |
| Cell Experiment | |
|---|---|
| Cell lines | LU1205 and WM35 cells |
| Preparation method | Apoptosis studies. Cells were exposed to soluble TRAIL (50 ng/mL) alone or in combination with cycloheximide (2 μg/mL). Different variants of combined treatment were used, including γ-irradiation (5 Gy), in the presence or absence of specific inhibitors of signaling pathways followed by TRAIL treatment. Apoptosis was then assessed by quantifying the percentage of hypodiploid nuclei using fluorescence-activated cell sorting analysis. |
| Concentrations | 10 μM |
| Incubation time | 48 h |
| Animal Experiment | |
|---|---|
| Animal models | LU1205 cells Human melanoma transplant in nude mice. |
| Formulation | DMSO |
| Dosages | 10 μM |
| Administration | |
| Molecular Weight | 395.49 |
| Formula | C21H17NO3S2 |
| CAS Number | 587871-26-9 |
| Solubility (25°C) | DMSO 39 mg/mL |
| Storage |
Powder -20°C 3 years ; 4°C 2 years In solvent -80°C 6 months ; -20°C 1 month |
| Related ATM/ATR Products |
|---|
| ART0380
ART0380 is a potent and selective ATR kinase inhibitor. |
| KU 59403
KU 59403 is a potent ATM inhibitor, with IC50 values of 3 nM, 9.1 μM and 10 μM for ATM, DNA-PK and PI3K, respectively. |
| SKLB-197
SKLB-197 showed an IC50 value of 0.013 μM against ATR but very weak or no activity against other 402 protein kinases. |
| ATR-IN-4
ATR-IN-4 is a potent ATR (Ataxia telangiectasia mutated gene Rad 3-associated kinase) inhibitor. |
| M3541
M3541 is a potent, ATP-competitive and selective ATM inhibitor with an IC50 of 0.25 nM. |
All AbMole products are for research use only, cannot be used for human consumption or veterinary use. We do not provide products or services to individuals. Please comply with the intended use and do not use AbMole products for any other purpose.


Products are for research use only. Not for human use. We do not sell to patients.
© Copyright 2010-2024 AbMole BioScience. All Rights Reserved.
