All AbMole products are for research use only, cannot be used for human consumption.
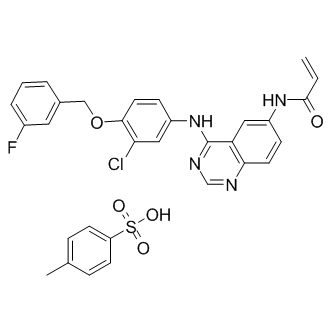
AST-1306 is a novel irreversible epidermal growth factor receptor inhibitor for EGFR and ErbB4 with IC50 of 0.5 and 0.8 nM, respectively. AST-1306 selectively inhibits the tyrosine kinase activities of ErbB2 with an IC50 of 3 nM. AST-1306 also potently prevents the EGFR T790M/L858R double mutant with an IC50 value of 12 nM. For this, AST-1306 is approximately 500-fold more potent than lapatinib. AST-1306 is more than 3000-fold selective for ErbB family kinases over other kinase families including PDGFR, KDR and c-Met. AST-1306 might covalently bind to specific amino acid residues of EGFR and ErbB2, AST-1306 significantly and concentration-dependently prevents the growth of HIH3T3-EGFR T790M/L858R cells. AST-1306 effectively suppresses EGFR phosphorylation in HIH3T3-EGFR T790M/L858R cells. Moreover, AST-1306 prevents the growth of NCI-H1975 cells expressing the EGFR T790M/L858R mutation in a concentration-dependent manner. AST-1306 blocks phosphorylation of EGFR and downstream pathways as well. In addition, AST-1306 dose-dependently and markedly inhibits EGF-induced EGFR phosphorylation in A549 cells. AST-1306 inhibits the phosphorylation of EGFR and ErbB2, and downstream signaling in human cancer cells including A549 cells, Calu-3 cells and SK-OV-3 cells. Twice daily oral administration of AST-1306 gives rise to a dramatic prevention of tumor growth in SK-OV-3 and Calu-3 xenograft models. In SK-OV-3 models, tumors almost completely disappears after treatment with AST-1306 for 7 days. In contrast, AST-1306 only slightly inhibits the growth of tumor in HO-8910 and A549 xenograft models. Therefore, the antitumor efficacy of AST-1306 is greater in ErbB2-overexpressing tumor models than in models expressing low levels of ErbB2. AST-1306 is well tolerate. In addition, oral administration of AST-1306 twice daily for 3 weeks dramatically suppresses the growth of tumor in the FVB-2/Nneu models. After treatment for 11 days, tumors almost completely disappears. The body weights of the mice reduces by less than 20% during treatment. AST-1306 has entered in a phase I clinical trial in the treatment of EGFR-overexpressing tumors.
| Cell Experiment | |
|---|---|
| Cell lines | Calu-3 and A-549 cell lines |
| Preparation method | Using the SRB (Sulforhodamine B) assay to evaluate cell (including Calu-3, A-549 cell line et al.) proliferation . Briefly, seeding cells into 96-well plates and grown for 24 hours. Then treating the cells with increasing concentrations of AST-1306 and grown for a further 72 hours. The medium remains unchanged until the completion of the experiment. Then fixing the cells with 10% precooled trichloroacetic acid (TCA) for 1 hour at 4 °C and staining for 15 min at room temperature with 100 μL of 4 mg/mL SRB solution in 1% acetic acid. Then removingthe SRB , and quickly rinsing the cells five times with 1% acetic acid. After cells are air-dried, protein-bound dye is dissolved in 150 μL of 10 mM Tris base for 5 min and measured at 515 nm using a multiwell spectrophotometer.Calculating the inhibition rate on cell proliferation as (1 - A515 treated/A515 control) × 100%. Obtaining the IC50 value by the Logit method and determining from the results of at least 3 independent tests. |
| Concentrations | 0.001-1 μM |
| Incubation time | 72 hours |
| Animal Experiment | |
|---|---|
| Animal models | Nude mice bearing SK-OV-3 xenograft tumors and SK-OV-3FVB-2/Nneu transgenic mouse |
| Formulation | 0.5% hydroxypropyl methylcellulose (HPMC), grinding with agate morta |
| Dosages | 25 mg/kg, 50 mg/kg and 100 mg/kg |
| Administration | p.o., twice daily |
| Molecular Weight | 621.08 |
| Formula | C24H18ClFN4O2 |
| CAS Number | 1050500-29-2 |
| Solubility (25°C) | DMSO 50 mg/mL |
| Storage |
Powder -20°C 3 years ; 4°C 2 years In solvent -80°C 6 months ; -20°C 1 month |
| Related EGFR/HER2 Products |
|---|
| Sevabertinib
Sevabertinib is an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor with antitumor activity. |
| BPIQ-I
BPIQ-I (PD 159121) is a potent and ATP-competitive EGFR tyrosine kinase inhibitor.. |
| HKI-357
HKI-357 is an irreversible dual inhibitor of EGFR and ERBB2 with IC50s of 34 nM and 33 nM, respectively. |
| SJF-1528
SJF-1528 is a potent EGFR PROTAC degrader with DC50 values of 39.2 nM and 736.2 nM for wild-type EGFR in OVCAR8 cells and Exon 20 Ins mutated EGFR in HeLa cells. |
| CH7233163
CH7233163 is a noncovalent ATP-competitive inhibitor for EGFR-Del19/T790M/C797S. |
All AbMole products are for research use only, cannot be used for human consumption or veterinary use. We do not provide products or services to individuals. Please comply with the intended use and do not use AbMole products for any other purpose.


Products are for research use only. Not for human use. We do not sell to patients.
© Copyright 2010-2024 AbMole BioScience. All Rights Reserved.
