All AbMole products are for research use only, cannot be used for human consumption.
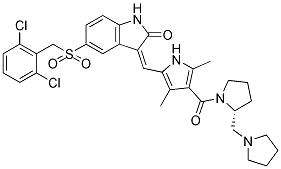
PHA-665752 is a small-molecule inhibitor of c-Met, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. c-Met is a tyrosine kinase receptor for hepatocyte growth factor/scatter factor (HGF/SF). PHA665752 inhibited specific phosphorylation of TPR-MET as well as phosphorylation of downstream targets of the mammalian target of rapamycin pathway. Short-term treatment with PHA-665752 decreased the numbers of premalignant lung lesions and induced apoptosis in tumor cells and vascular endothelial cells within lesions. In cell culture, PHA-665752 induced apoptosis of a lung adenocarcinoma cell line derived from Kras(LA1) mice (LKR-13) and a murine lung endothelial cell line (MEC). PHA665752 was shown to inhibit cMet/HGF/SF signaling in vitro, suggesting c-Met inhibitors may have efficacy for blocking local progression and/or metastatic spread of c-Met-positive NBL in vivo.
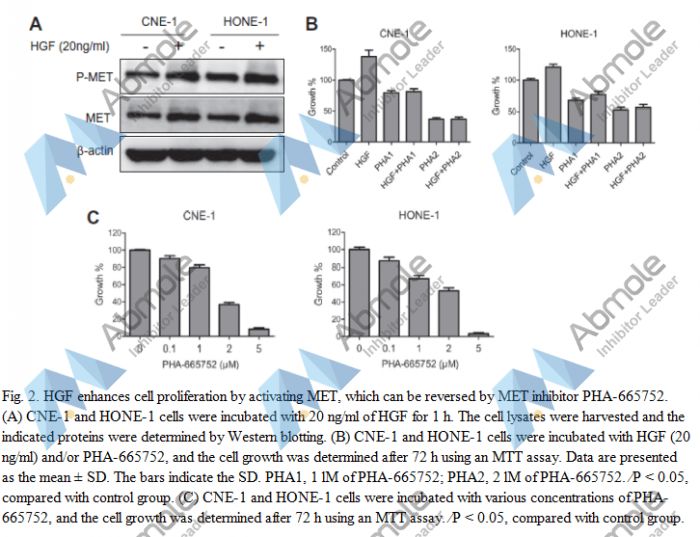 |
Source | Biochem Biophys Res Commun (2014). Figure 2.PHA-665752 |
| Method | MTT assay | |
| Cell Lines | CNE-1 and HONE-1 cells | |
| Concentrations | 2 μM | |
| Incubation Time | 72 h | |
| Results | Further experiments showed that PHA-665752 inhibited cell proliferation in a dose-dependent manner in the CNE-1 and HONE-1 cells |
| Cell Experiment | |
|---|---|
| Cell lines | S114, GTL-16, NCI-H441, or BxPC-3 cells |
| Preparation method | Cell Proliferation Assays. S114, GTL-16, NCI-H441, or BxPC-3 cells were seeded in 96-well plates at 9000 cells/well in medium with 10% FBS. After incubation for 48 h in low serum (0.5% FBS, S114; 0.1% FBS, GTL-16, NCI-H441, and BxPC-3), cells were treated with different concentrations of PHA-665752 for 18 h at 37°C. HGF (50 ng/ml) was added for 18 h before BrdUrd for studies involving GTL-16, H441, and BxPC-3. After incubation with BrdUrd labeling reagent for 1 h, cells were fixed and BrdUrd incorporation into newly synthesized DNA was assessed using anti-BrdUrd peroxidase-conjugated antibody followed by colorimetric determination at 630 nm. |
| Concentrations | 0~1.25µM |
| Incubation time | 18h |
| Animal Experiment | |
|---|---|
| Animal models | athymic mice bearing S114 or GTL-16 tumor xenografts |
| Formulation | L-lactate (pH 4.8) and 10% polyethylene glycol |
| Dosages | 7.5, 15, 30 mg/kg/day |
| Administration | i.v. bolus via tail vein injection |
| Molecular Weight | 641.61 |
| Formula | C32H34Cl2N4O4S |
| CAS Number | 477575-56-7 |
| Solubility (25°C) | DMSO 60 mg/mL |
| Storage |
Powder -20°C 3 years ; 4°C 2 years In solvent -80°C 6 months ; -20°C 1 month |
| Related c-Met Products |
|---|
| Zurletrectinib
Zurletrectinib (ICP-723) is a potent tyrosine kinase inhibitor. Zurletrectinib serves as an antineoplastic agent. Zurletrectinib can used for research of TRK-mediated related diseases. |
| ABN401
ABN401 is a highly potent and selective ATP-competitive c-MET inhibitor with an IC50 value of 10 nM. |
| (Z)-Semaxanib
(Z)-Semaxanib is a potent tyrosine kinase inhibitor. |
| MET kinase-IN-2
MET kinase-IN-2 is a potent, selective, orally bioavailable MET kinase inhibitor with an IC50 of 7.4 nM. |
| BMS-817378
BMS-817378 is a potent and selective inhibitor of MET with IC50 of 1.7 nM. |
All AbMole products are for research use only, cannot be used for human consumption or veterinary use. We do not provide products or services to individuals. Please comply with the intended use and do not use AbMole products for any other purpose.


Products are for research use only. Not for human use. We do not sell to patients.
© Copyright 2010-2024 AbMole BioScience. All Rights Reserved.
