All AbMole products are for research use only, cannot be used for human consumption.
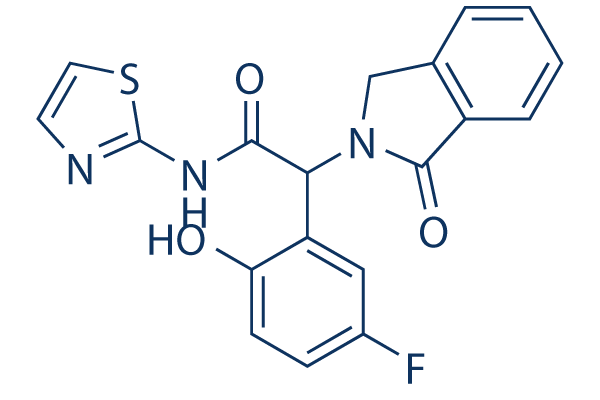
EAI045 is a fourth generation allosteric inhibitor of mutant EGFR, which inhibits EGFR at 10 μM ATP, EGFRL858R, EGFRT790M and EGFRL858R/T790M with IC50 values of 1.9, 0.019, 0.19 and 0.002 μM respectively.
| Cell Experiment | |
|---|---|
| Cell lines | H1975, H3255 and HaCaT cell lines |
| Preparation method | H1975, H3255 and HaCaT cell lines are plated in solid white 384-well plates at 500 cells per well in 10% FBS RPMI penicillin/streptomycin media. Using a Pin Tool, 50 nl of serial diluted compounds are transferred to the cells. After 3 days, cell viability is measured. |
| Concentrations | Serial dilution (0-100 μM) |
| Incubation time | 3 days |
| Animal Experiment | |
|---|---|
| Animal models | EGFR(TL) (bearing L858R/T790M point mutations) and EGFR(TD) (bearing exon19del/T790M point mutations) mice |
| Formulation | 10% NMP (10% 1-methyl-2-pyrrolidinone:90% PEG-300) |
| Dosages | 60 mg/kg |
| Administration | oral gavage |
| Molecular Weight | 383.40 |
| Formula | C19H14FN3O3S |
| CAS Number | 1942114-09-1 |
| Solubility (25°C) | 76 mg/mL in DMSO |
| Storage |
Powder -20°C 3 years ; 4°C 2 years In solvent -80°C 6 months ; -20°C 1 month |
| Related EGFR/HER2 Products |
|---|
| Sevabertinib
Sevabertinib is an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor with antitumor activity. |
| BPIQ-I
BPIQ-I (PD 159121) is a potent and ATP-competitive EGFR tyrosine kinase inhibitor.. |
| HKI-357
HKI-357 is an irreversible dual inhibitor of EGFR and ERBB2 with IC50s of 34 nM and 33 nM, respectively. |
| SJF-1528
SJF-1528 is a potent EGFR PROTAC degrader with DC50 values of 39.2 nM and 736.2 nM for wild-type EGFR in OVCAR8 cells and Exon 20 Ins mutated EGFR in HeLa cells. |
| CH7233163
CH7233163 is a noncovalent ATP-competitive inhibitor for EGFR-Del19/T790M/C797S. |
All AbMole products are for research use only, cannot be used for human consumption or veterinary use. We do not provide products or services to individuals. Please comply with the intended use and do not use AbMole products for any other purpose.


Products are for research use only. Not for human use. We do not sell to patients.
© Copyright 2010-2024 AbMole BioScience. All Rights Reserved.
