All AbMole products are for research use only, cannot be used for human consumption.
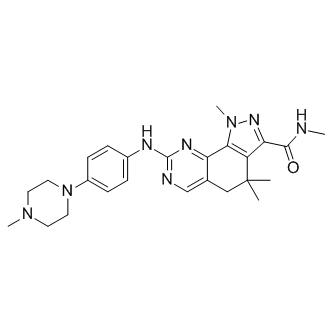
PHA-848125 is a potent ATP-competitive cyclin A/CDK2 inhibitor with an IC50 of 45 nM. HA-848125 inhibits, although with lower potency, the activities of cyclin H/CDK7, cyclin D1/CDK4, p35/CDK5, as well as cyclin E/CDK2 and cyclin B/CDK1 with IC50 of 0.15, 0.16, 0.265, 0.363, 0.398 μM, respectively.Besides, thropomyosin receptor kinase A is also prevented by PHA-848125 in the same nanomolar range of CDKs. In the most PHA-848125-sensitive cell line, PHA-848125 induces a concentration-dependent G (1) arrest.PHA-848125 also impairs phosphorylation of the retinoblastoma protein at CDK2 and CDK4 specific sites, reduces retinoblastoma protein and cyclin A levels, and increases p21 (Cip1), p27 (Kip1) and p53 expression. PHA-848125 is added to the cells 48 hours after TMZ and cell growth is evaluated after 3 additional days of culture.Drug combination of TMZ, BG and PHA-848125 produces additive or synergistic effects on cell growth, depending on the cell line. When TMZ plus BG are used in combination with PHA-848125 against cultured normal melanocytes, neither synergistic nor additive antiproliferative effects are observed.In the preclinical xenograft A2780 human ovarian carcinoma model PHA-848125 reveals good efficacy and is well tolerated upon repeated daily treatments. Treatment of K-Ras (G12D)LA2 mice with 40 mg/kg twice daily for 10 days with PHA-848125 blocks the growth of a significant tumor at the end of treatment and is accompanied by a reduction in the cell membrane turnover. On the other hand, PHA-848125 posseseese significant antitumor activity in various human xenografts, carcinogen-induced tumors and in disseminated primary leukemia models.

Fish Physiol Biochem. 2019 Dec;45(6):1829-1843.
A novel CDK-2 homolog identified in lamprey, Lampetra japonica, with roles in apoptosis.
PHA-848125 purchased from AbMole
| Cell Experiment | |
|---|---|
| Cell lines | Melanoma cells |
| Preparation method | Suspending melanoma cells in culture media at a concentration of 2 × 104 cells/mL, dispensing in 50 μL aliquots into flat-bottom 96-well plates and allowed to adhere overnight at 37 °C. Then adding Graded amounts of PHA-848125 or TMZ to the wells (4 wells per point) in 50 μL of CM and incubating the plates at 37 °C in a 5% CO2 humidified atmosphere for 5 days. The cytotoxic effects of TMZ are also evaluated in combination with the MGMT inhibitor BG. To this end, adding 10 μM BG to the plates 2 hours before TMZ and left in culture for the entire period of cell exposure to the drug. ContS1017rol groups are represented by untreated cells and cells treated with BG or DMSO alone. The growth of the cells treated with BG or DMSO alone does not differ from that of untreated cells. MGMT activity of BG-treated cells is undetectable 2 hours after the addition of the inhibitor PHA-848125 and remained essentially undetectable up to the end of the assay. Normal melanocytes are suspended in MGM at the concentration of 1.6 × 105 cells/mL, plated (50 μL/well) and exposed to TMZ + BG or to PHA-848125 as described for melanoma cells. At the end of the incubation period, using the MTT assay to evaluate cell growth . Briefly, 0.1 mg of MTT (in 20 μL of PBS) is added to each well and cells are incubated at 37 °C for 4 hours. Cells are then lysed with a buffer (0.1 mL/well) containing 20% SDS and 50% N,N-dimethylformamide, pH 4.7. After overnight incubation, the absorbance is read at 595 nm using a 3550-UV microplate reader. Cell sensitivity to drug treatment is expressed in terms of IC50 (drug concentration producing 50% inhibition of cell growth, calculated on the regression line in which absorbance values at 595 nm are plotted against the logarithm of drug concentration). |
| Concentrations | 5.7 mg/mL |
| Incubation time | 2 hours |
| Animal Experiment | |
|---|---|
| Animal models | K-Ras(G12D)LA2 mice |
| Formulation | 5% dextrose solution |
| Dosages | 40 mg/kg twice daily for 10 days |
| Administration | Oral administration |
| Molecular Weight | 460.57 |
| Formula | C25H32N8O |
| CAS Number | 802539-81-7 |
| Solubility (25°C) | DMSO 90 mg/mL |
| Storage |
Powder -20°C 3 years ; 4°C 2 years In solvent -80°C 6 months ; -20°C 1 month |
| Related CDK Products |
|---|
| (E/Z)-Zotiraciclib citrate
(E/Z)-Zotiraciclib citrate is a potent CDK2, JAK2, and FLT3 inhibitor. |
| Abemaciclib metabolites M2
Abemaciclib metabolite M2 (LSN2839567) is a metabolite of Abemaciclib, acts as a potent CDK4 and CDK6 inhibitor, with IC50 values of 1.2 and 1.3 nM, respectively. |
| CDK9-IN-1
CDK9-IN-1 is a novel, selective CDK9 inhibitor, with an IC50 of 39 nM for CDK9/CycT1, it can be used for the research of HIV infection. |
| XO44
XO44 (PF-6808472) is a broad-spectrum covalent kinase probe. |
| ML 315
ML 315 is a selective dual inhibitor of CDK and DYRK with IC50s of 68 nM and 282 nM, respectively. |
All AbMole products are for research use only, cannot be used for human consumption or veterinary use. We do not provide products or services to individuals. Please comply with the intended use and do not use AbMole products for any other purpose.


Products are for research use only. Not for human use. We do not sell to patients.
© Copyright 2010-2024 AbMole BioScience. All Rights Reserved.
