In vitro: MK-8617 is not a significant inhibitor of the cytochrome p450 enzymes in vitro (IC50), CYP1A2, 3A4, 2B6, 2C9, 2C19, or 2D6, >60 μM, and is a moderate reversible inhibitor of CYP2C8 at 1.6 μM in vitro. MK-8617 is inactive when screened at 10 μM against a general panel of 171 radioligand binding and enzymatic assays. In vivo: Tritiated MK-8617 exhibits minimal metabolic turnover in liver microsomes (+NADPH) from rat, dog, and monkey (<10% turover) but significant turnover in human liver microsomes (34% turnover) after 60 min (10 μM compound, 1 mg/mL microsomal protein). In terms of its pharmacokinetic profile, MK-8617 shows good oral bioavailability across species (36−71%), with low clearance and volume of distribution. The compound still has a relatively long elimination half-life across preclinical species. In mice (C57Bl/6), single doses of 5 and 15 mpk po (n = 3) causes increases in circulating reticulocytes measured on both 3 and 4 days postcompound challenge. In rat (Sprague−Dawley), a single dose titration of 1.5, 5, and 15 mpk po (n = 5) causes a large increase in serum erythropoietin(EPO) levels of 1.7-, 8-, and 204-fold relative to vehicle, respectively. Increases in circulating reticulocytes are observed at 5 and 15 mg/kg 3 days after challenge and, with the 15 mg dose, at 4 days after challenge.
| Cell Experiment | |
|---|---|
| Cell lines | |
| Preparation method | |
| Concentrations | |
| Incubation time | |
| Animal Experiment | |
|---|---|
| Animal models | C57Bl/6 mice |
| Formulation | DMSO/PEG400/water (5:40:55, v/v/v) |
| Dosages | 1 mg/kg po, 0.5 mg/kg iv |
| Administration | i.v or p.o |
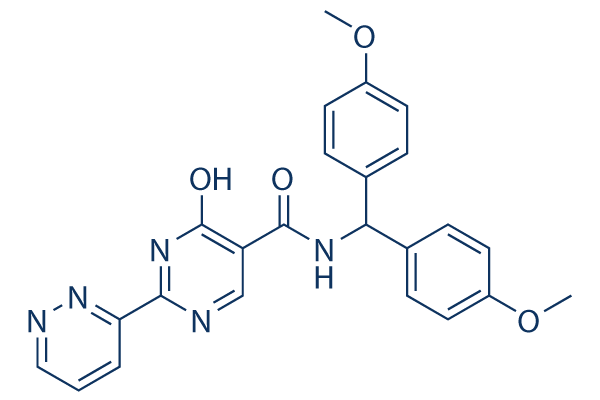
| Molecular Weight | 443.45 |
| Formula | C24H21N5O4 |
| CAS Number | 1187990-87-9 |
| Solubility (25°C) | 88 mg/mL in DMSO |
| Storage |
Powder -20°C 3 years ; 4°C 2 years In solvent -80°C 6 months ; -20°C 1 month |
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of Compound A used for a mouse (20 mg/kg) to a dose based on the BSA for a rat, multiply 20 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for Compound A of 10 mg/kg.
| Related HIF Products |
|---|
| 3-Aminobenzoic acid
3-Aminobenzoic acid is a hypoxia-inducible factor-1α (HIF-1α) inhibitor. |
| Steppogenin
Steppogenin is a potent inhibitor of HIF-1α and DLL4, with IC50 values of 0.56 and 8.46 μM, respectively. |
| HIV-IN-7
Axl-IN-16 is a Axl inhibitor. |
| HIF-2α-IN-9
HIF-2α-IN-9 is an inhibitor ofHIF-2α. |
| PHD-IN-2
PHD-IN-2 is a PHD antagonist (IC50: < 5 nM). |


Products are for research use only. Not for human use. We do not sell to patients.
© Copyright 2010-2023 AbMole BioScience. All Rights Reserved.
