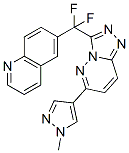
JNJ-38877605 is a small-molecule, ATP-competitive inhibitor of the catalytic activity of c-Met. JNJ-38877605 showed f600-fold selectivity for c-Met compared with a panel of f250 diverse tyrosine and serine-threonine kinases and was found to potently inhibit HGF-stimulated and constitutively activated c-Met phosphorylation in vitro. In addition, JNJ-38877605 induces regression of U87-MG xenografts in vivo.
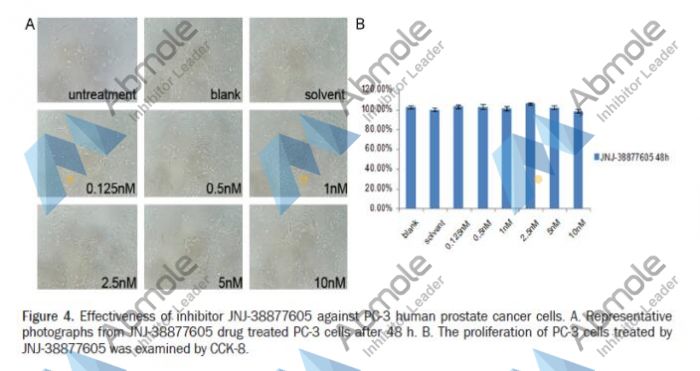 |
Source | Int J Clin Exp Med (2015). Figure 4. JNJ-38877605 |
| Method | CCK-8 assay | |
| Cell Lines | PC-3 human prostate cancer cells | |
| Concentrations | 0.125 nM, 0.5 nM, 1 nM, 2.5 nM, 5 nM, 10 nM | |
| Incubation Time | 48 h | |
| Results | However, different concentrations of saracatinib, linsitinib and JNJ-38877605 did not inhibit PC-3 cells proliferation after 48 h, high or low concentrations of the three inhibitors did not inhibit PC-3 cells proliferation at all. |
| Cell Experiment | |
|---|---|
| Cell lines | S114, GTL-16, NCI-H441, or BxPC-3 cells |
| Preparation method | Cell Proliferation Assays. S114, GTL-16, NCI-H441, or BxPC-3 cells were seeded in 96-well plates at 9000 cells/well in medium with 10% FBS. After incubation for 48 h in low serum (0.5% FBS, S114; 0.1% FBS, GTL-16, NCI-H441, and BxPC-3), cells were treated with different concentrations of PHA-665752 for 18 h at 37°C. HGF (50 ng/ml) was added for 18 h before BrdUrd for studies involving GTL-16, H441, and BxPC-3. After incubation with BrdUrd labeling reagent for 1 h (Sigma Biochemicals, St. Louis, MO), cells were fixed and BrdUrd incorporation into newly synthesized DNA was assessed using anti-BrdUrd peroxidase-conjugated antibody followed by colorimetric determination at 630 nm. |
| Concentrations | 0~1.25µM |
| Incubation time | 18 h |
| Animal Experiment | |
|---|---|
| Animal models | Female athymic mice bearing S114 or GTL-16 tumor xenografts |
| Formulation | L-lactate (pH 4.8) and 10% polyethylene glycol |
| Dosages | 25 mg/kg a single i.v. dose |
| Administration | via bolus i.v. tail vein injection |
| Molecular Weight | 377.35 |
| Formula | C19H13F2N7 |
| CAS Number | 943540-75-8 |
| Solubility (25°C) | DMSO 30 mg/mL |
| Storage |
Powder -20°C 3 years ; 4°C 2 years In solvent -80°C 6 months ; -20°C 1 month |
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of Compound A used for a mouse (20 mg/kg) to a dose based on the BSA for a rat, multiply 20 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for Compound A of 10 mg/kg.
| Related c-Met Products |
|---|
| BMS-817378
BMS-817378 is a potent and selective inhibitor of MET with IC50 of 1.7 nM. |
| Capmatinib dihydrochloride hydrate
Capmatinib dihydrochloride hydrate is a potent, orally active, selective, ATP-competitive c-Met kinase inhibitor (IC50=0.13 nM) that inhibits the phosphorylation of c-MET, as well as downstream effector proteins of the c-MET pathway, such as ERK1/2, AKT, FAK, In addition, Capmatinib dihydrochloride hydrate effectively inhibited the proliferation and migration of c-Met-dependent tumor cells, induced apoptosis, and demonstrated antitumor activity in a mouse model of tumor. Capmatinib dihydrochloride hydrate is mainly metabolized by CYP3A4 and aldehyde oxidase. |
| Caveolin-1 (82-101) amide (human, mouse, rat)
Caveolin-1 (82-101) amide (human, mouse, rat) (Caveolin-1 scaffolding domain peptide) is a peptide that reverses aging-associated deleterious changes in multiple organs. |
| Norleual
Norleual, an angiotensin (Ang) IV analog, is a hepatocyte growth factor (HGF)/c-Met inhibitor with an IC50 of 3 pM. |
| Fosgonimeton
Fosgonimeton is a potentially first-in-class Hepatocyte Growth Factor Receptor (HGF) agonist that enhances HGF/MET signaling pathway activity for Alzheimer's Disease (AD) research. |


Products are for research use only. Not for human use. We do not sell to patients.
© Copyright 2010-2023 AbMole BioScience. All Rights Reserved.
